

What is sickle cell disease?
Sickle cell disease refers to a group of inherited health conditions that affect red blood cells. The specific type a person has depends on which hemoglobin gene mutations (changes) they inherited from their parents. The hemoglobin gene gives instructions for a part of hemoglobin, which is the protein that travels through the blood to deliver oxygen throughout the body. The most serious form of this health condition is known as sickle cell anemia (SS).
Who is most affected?
Sickle cell disease is most common in people with ancestry from Africa, South or Central America (particularly Panama), the Caribbean islands, Mediterranean nations (such as Turkey, Greece, and Italy), India, and Saudi Arabia.
We do not have information on the exact number of children born with sickle cell disease worldwide. This is because, unlike in Europe and North America, less economically developed countries, which likely have the most cases, do not screen newborn babies for sickle cell disease. However, based on global birth rates and data on how often individuals carry the gene for sickle cell disease, it is estimated that around 312,000 children are born with sickle cell disease type SS each year. This estimate includes about 300 births in the UK and almost 3000 in the USA.
How is sickle cell disease inherited?
Genes come in pairs, you get one set from your mother and one set from your father. To have sickle cell disease at birth, a child needs to get a copy of the sickle cell gene from both parents. This usually happens when both parents are “carriers” of the sickle cell gene. In this case, there’s a 1 in 4 chance that the child will receive copies of the sickle cell gene from both parents and be born with the disease. Alternatively, it can also happen when one parent has sickle cell disease, and the other is a carrier.
People with all types of sickle cell disease inherit the gene for hemoglobin S from 1 parent and a gene for another type of hemoglobin from 1 parent. Hemoglobin S (also called sickle hemoglobin) is the most common type of abnormal hemoglobin. Normal red blood cells are flexible and disc-shaped, but in sickle cell disease they can become stiff, rigid, and shaped like a crescent or sickle because the hemoglobin inside them clumps together. Sickle red blood cells do not live as long as healthy blood cells and can cause an increase in blood viscosity (thickness) as well as blockages in blood flow inside blood vessels in any organ, including the brain.
Sickle cell disease affects both children and adults, but the initial symptoms typically show up in the first year of life. Managing sickle cell disease is a lifelong commitment that involves continuous, adaptive care.
Why it’s important to raise awareness about stroke in sickle cell disease:
Stroke is one of the most common and serious problems in sickle cell disease. It can lead to problems with movement, speech, and thinking and may even cause death.
Risk of stroke
There is about 250 times higher occurrence of stroke in sickle cell disease compared to the general pediatric population. Without screening for stroke risk and treatments to lower this risk (also called primary stroke prevention), around 5 to 10% of children with sickle cell disease will experience a stroke, most commonly in the first ten years of life. Strokes tend to happen again in most children with sickle cell disease who do not receive preventative measures. For this reason, current efforts aim to prevent both the first (primary) and recurring (secondary) strokes in this high-risk population.
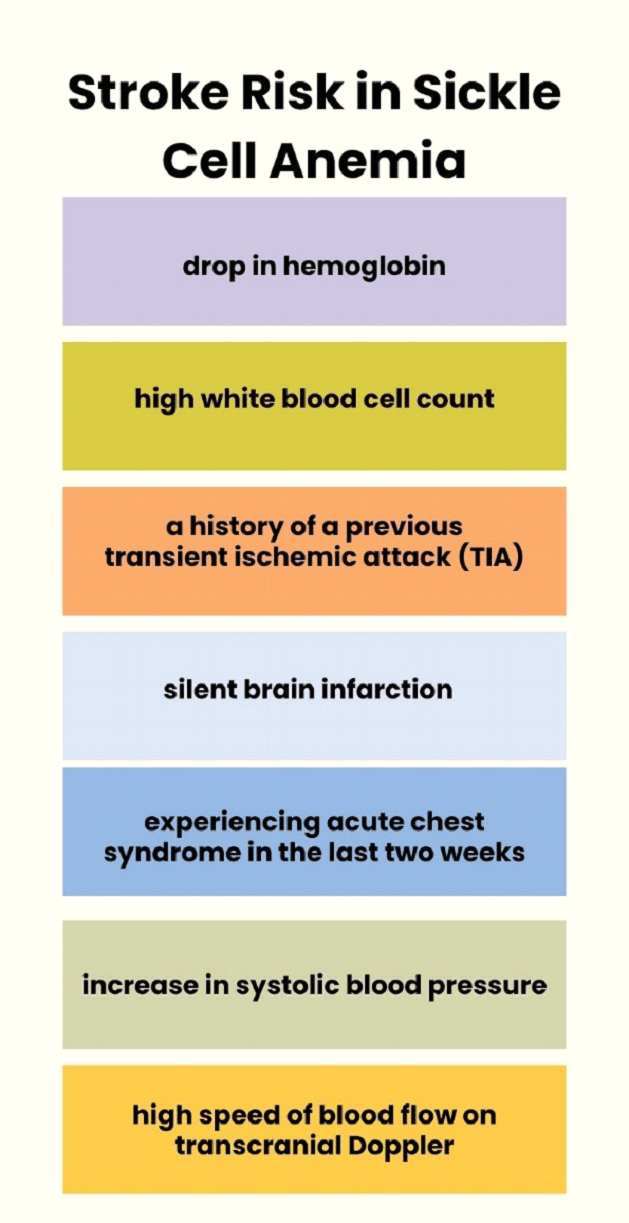
Risk of first stroke in patients with sickle cell disease type SS:
There are many factors associated with an increased risk of experiencing a stroke in a patient with SCD. These include a drop in hemoglobin, a high white blood cell count, a history of a previous transient ischemic attack (TIA), silent brain infarction, experiencing acute chest syndrome in the last two weeks, an increase in systolic blood pressure, and a high speed of blood flow on transcranial Doppler.
How does using transcranial Doppler for screening affect the risk of stroke?
Transcranial Doppler is a tool that allows us to see the major blood vessels inside the skull to find blood flowing very quickly. The fast flowing blood in arteries to the brain is a sign that a child has a higher risk of stroke. The measurement obtained from this tool can be in one of three categories: normal (less than 170 cm/sec) , abnormal (≥200 cm/sec), or conditional if the measurement is not normal but did not reach abnormal level (170–199 cm/sec). Without taking any medical action, people with conditional speeds have a 1–3% chance of having a stroke every year, while those with abnormal speeds have a stroke risk as high as 10% each year. Transcranial Doppler screening should start at age two and continue once each year until age sixteen if the speed of the flow of blood is normal, every 3 to 6 months if they’re conditional, or every month if they’re abnormally high.

How to protect children with sickle cell disease from getting stroke (primary prevention)
Children with abnormal transcranial Doppler measurements should receive regular blood transfusion therapy. This treatment reduces the risk of stroke by more than 90% compared to children with abnormal transcranial Doppler measurements who do not receive any therapy. After one year of regular blood transfusion therapy, children may switch from transfusions to a medication, hydroxyurea, if the child does not have narrow blood vessels in the brain.
How patients present with acute stroke
When children with sickle cell disease have a stroke, they may show focal neurological deficits, which include sudden weakness or numbness on one side of the face and body, difficulty talking, trouble seeing, or problems with balance. Other possible symptoms include headaches, sleepiness, seizures, difficulty in understanding or expressing words (aphasia), or cognitive decline as poor academic performance and attentional deficits.
What is acute management?
Children with sickle cell disease who have a focal neurologic deficit need a comprehensive care approach coordinated by hematology teams. Acute management involves immediate assessment by both a hematologist and neurologist, with a collaborative discussion of the assessment and treatment plan.
Recommended imaging approach
For people presenting to the emergency room with a suspected stroke, often a CT of the brain will be done to check for a hemorrhagic (bleeding) stroke. After the patient stabilizes an MRI and MRV of the brain is recommended. If an MRI can be completed rapidly as part of the initial evaluation and the scan sequence has been set up to detect cerebral hemorrhage and thrombosis then a CT may not be needed.
Supportive care measures
Supportive care measures are things doctors can do to support a patient through a disease (as opposed to treating the disease). In children with sickle cell disease, this includes giving oxygen to the patient to keep oxygen saturation above 95%, using fever-reducing medications (antipyretics) and antibiotics if there’s a suspected infection, ensuring proper hydration, and addressing low blood pressure.
Goal of transfusion in sickle cell disease:
Blood transfusion with hemoglobin S-negative blood (meaning healthy blood cells that do not have the hemoglobin mutation) reduces the proportion of circulating sickle red blood cells in the body. This in turn decreases blood viscosity (thickness) and injury to blood vessels and red blood cells. It also increases the oxygen in the blood to help more oxygen get to the tissues.
Transfusion approach in acute stroke
When a child with sickle cell disease has a stroke, the doctors will give a blood transfusion. This could be a “simple transfusion” which means the child is given normal RBCs, without removal of sickle RBCs. Or, it could be an “exchange transfusion” which includes removal sickle RBCs while giving the child healthy RBCs. It’s worth noting that exchange transfusion has been linked to a lower risk of subsequent stroke compared to simple transfusion when presented at the time of a stroke.
Sub-acute care strategy following an acute stroke:
After discharge from the hospital, a child with a stroke should follow-up with a neurologist for a good neurological exam. Additionally, it would be good for the child to see a psychologist for testing to identify cognitive strengths and weaknesses, and to assess if there’s a need for accommodation at school or in the workplace.
Secondary stroke prevention
Blood transfusions can also be used to prevent a recurrent stroke. Stem cell transplants, that help the body produce healthy red blood cells, is another option. Without interventions such as regular blood transfusion, or stem cell transplantation, the chance of suffering another stroke after a first stroke is 50-90%. A daily medication called hydroxyurea is another option for children who cannot get long-term blood transfusions or stem cell transplants. Fortunately, these stroke prevention treatments work well to prevent stroke and keep a child’s brain healthy.
Resources
- https://www.nhs.uk/conditions/sickle-cell-disease/
- Chakravorty S, Williams TN. Sickle cell disease: a neglected chronic disease of increasing global health importance Archives of Disease in Childhood 2015;100:48-53. PMID: 25239949
- Jordan L, Swerdlow P, Coates TD. Systematic review of transition from adolescent to adult care in patients with sickle cell disease. J Pediatr Hematol Oncol. 2013 Apr;35(3):165-9. PMID: 23511487
- Adams RJ. Stroke prevention and treatment in sickle cell disease. Arch Neurol. 2001 Apr;58(4):565-8. PMID: 11295986
- Lee MT, Piomelli S, Granger S, Miller ST, Harkness S, Brambilla DJ, Adams RJ; STOP Study Investigators. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood. 2006 Aug 1;108(3):847-52. PMID: 16861341
- Ware RE, Helms RW; SWiTCH Investigators. Stroke With Transfusions Changing to Hydroxyurea (SWiTCH). Blood. 2012 Apr 26;119(17):3925-32. PMID: 22318199.
- Hulbert ML, Scothorn DJ, Panepinto JA, Scott JP, Buchanan GR, Sarnaik S, Fallon R, Chu JY, Wang W, Casella JF, Resar L, Berman B, Adamkiewicz T, Hsu LL, Smith-Whitley K, Mahoney D, Woods G, Watanabe M, DeBaun MR. Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anemia. J Pediatr. 2006 Nov;149(5):710-2. PMID: 17095350
- American Society of Hematology Clinical Practice Guidelineson Sickle Cell Disease
- Inforgraphic: Sickle Celll Disease and Pediatric Stroke
- Inforgraphic: Sickle Celll Disease and Pediatric Stroke
About the Author

Dr Fatma Ebeid
Professor of Pediatric Hematology Oncology and BMT Executive Director of MASRI- CRC
Cairo, Egypt
Dr Fatma Ebeid, professor of Pediatric Hematology Oncology and Bone Marrow Transplant and the executive director of Faculty of Medicine Ain Shams Research Institute-Clinical Research Center (MASRI- CRC). Dr. Ebeid has almost nineteen years of experience in the field of Pediatric Hematology in one of the largest referral centers in MENA region, she has special interest in inherited anemias and bone marrow failure disorders especially SCD. Currently she is the physician who directs the SCD clinic;she implemented the first center in Egypt that provides automated blood exchange transfusions for SCD patients. Dr Ebeid has participated in many international clinical trials addressing SCD ranging from iron chelation to anti-sickling investigational drugs and has a rich local database including pediatric and adult SCD patients. Dr. Ebeid has many international and national scientific publications, and she is a member of and has collaborated with national and international professional bodies, research groups and non-profit organizations.
Junior Editor: Natalie Mahgerefteh



{kind=link}